- Guides and Tutorials
- Bioconductor / RStudio
- Starting RStudio
- Using R / Bioconductor in AnVIL
- The R / Bioconductor AnVIL Package
- Running a Workflow
- Single-cell RNASeq with 'Orchestrating Single Cell Analysis' in R / Bioconductor
- Using AnVIL for teaching R / Bioconductor
- Reproducible Research with AnVILPublish
- Participant Stories
- Galaxy
- Starting Galaxy
- Jupyter
- Starting Jupyter
Running a Workflow
How to configure and run a workflow, based on the Bioconductor-Workflow-DESeq2 workspace. The workflow starts with FASTQ files and transforms them using salmon to the inputs required for Bioconductor DESeq2 analysis of differential expression.
Learning Objectives
This week we'll configure and run a workflow, based on the Bioconductor-Workflow-DESeq2 workspace (access required via course registration or email to mtmorgan.bioc@gmail.com). The workspace allows a complete bulk RNASeq differential expression analysis. The workflow starts with FASTQ files and transforms them using salmon to the inputs required for Bioconductor DESeq2 analysis of differential expression. Notebooks describe how the workspace was set up (so that it can be tailored to individual analyses) and also how the outputs of a successful workflow can be marshaled as inputs to an interactive DESeq2 analysis.
Key Resources
- Visit https://anvil.terra.bio to use the AnVIL platform.
- The salmon website and excellent DESeq2 vignette form the foundation for the workspace.
- Completed workflows are run in the cloud, but components are often developed locally using Cromwell. Workflows are written in the Workflow Description Language (WDL).
- Workflow tasks often make use of docker to create containers with the software necessary for the task.
Review
Previously...
- The course schedule contains links and videos of previous sessions.
Essential steps
- Login
- Workspaces
- Billing accounts
- (R-based) Jupyter notebooks or RStudio for interactive analysis
- Workflows for large-scale data processing
Cloud computing environment
- Runtime and persistent disk.
- Workspace DATA and buckets.
- AnVIL package for interaction with workspace components.
FAQ answers
- We upload workflows through GitHub / Dockstore, but also the Broad Methods Repository (YouTube); see also the WDL Puzzles workspace.
- Default name and namespace -- the runtime starts in a particular workspace, and the runtime knows the default namespace and name. So by default, I had
> avworkspace() [1] "deeppilots-bioconductor-may3/Bioconductor-Workshop-PopUp-mtmorgan" gsutil_cp(): CommandException: Downloading this composite object requires integrity checking with CRC32c, but your crcmod installation isn’t using...This is a bug that will be fixed in the underlying image for the runtime.
Workshop Activities
Setup & Tour
Setup
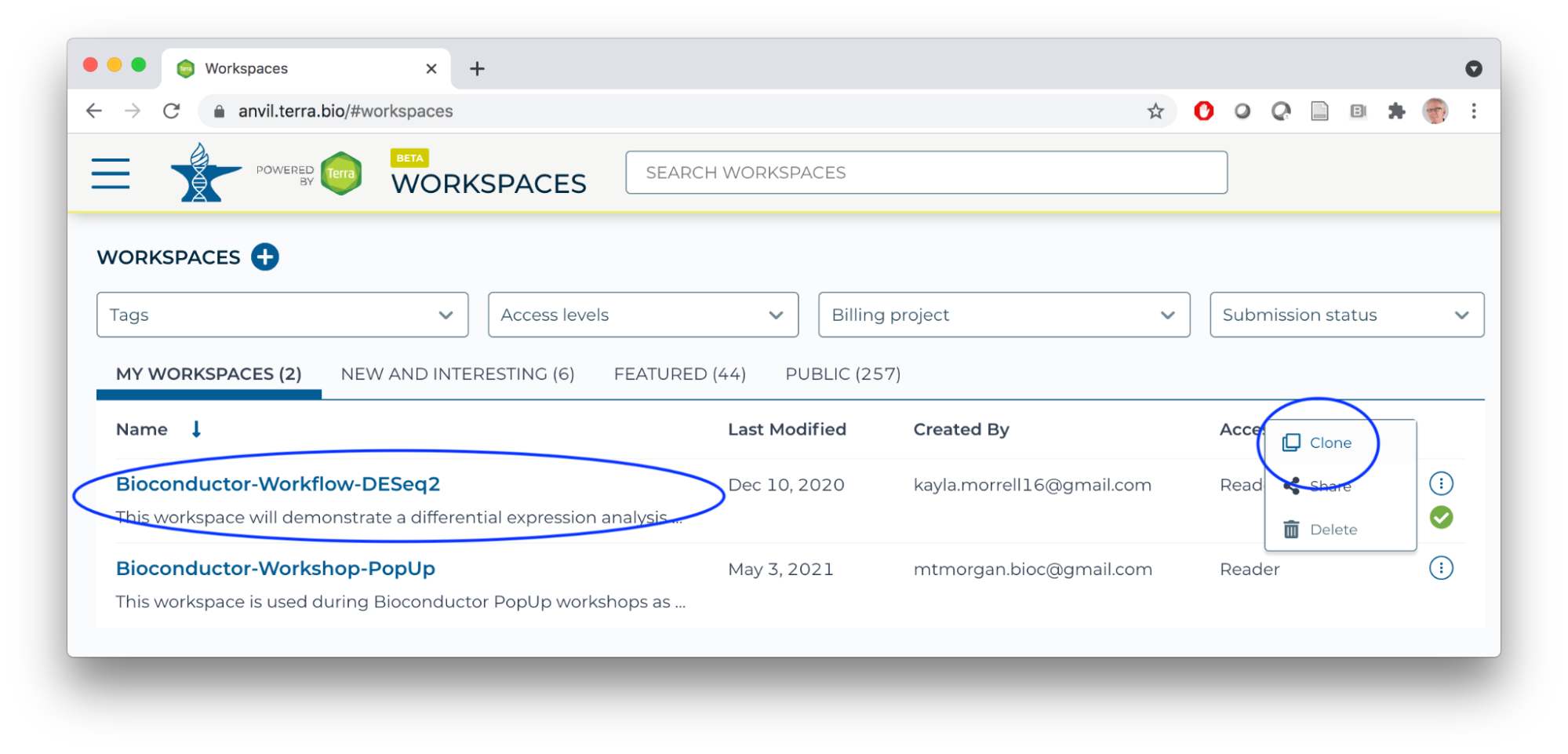
- Log in to AnVIL using the email address you used to register for the course and navigate (via the HAMBURGER) to Workspaces.
- Clone the Bioconductor-Workflow-DESeq2 workspace
- Unique workspace name
- Billing project: deeppilots-bioconductor-may10
Workspace tour
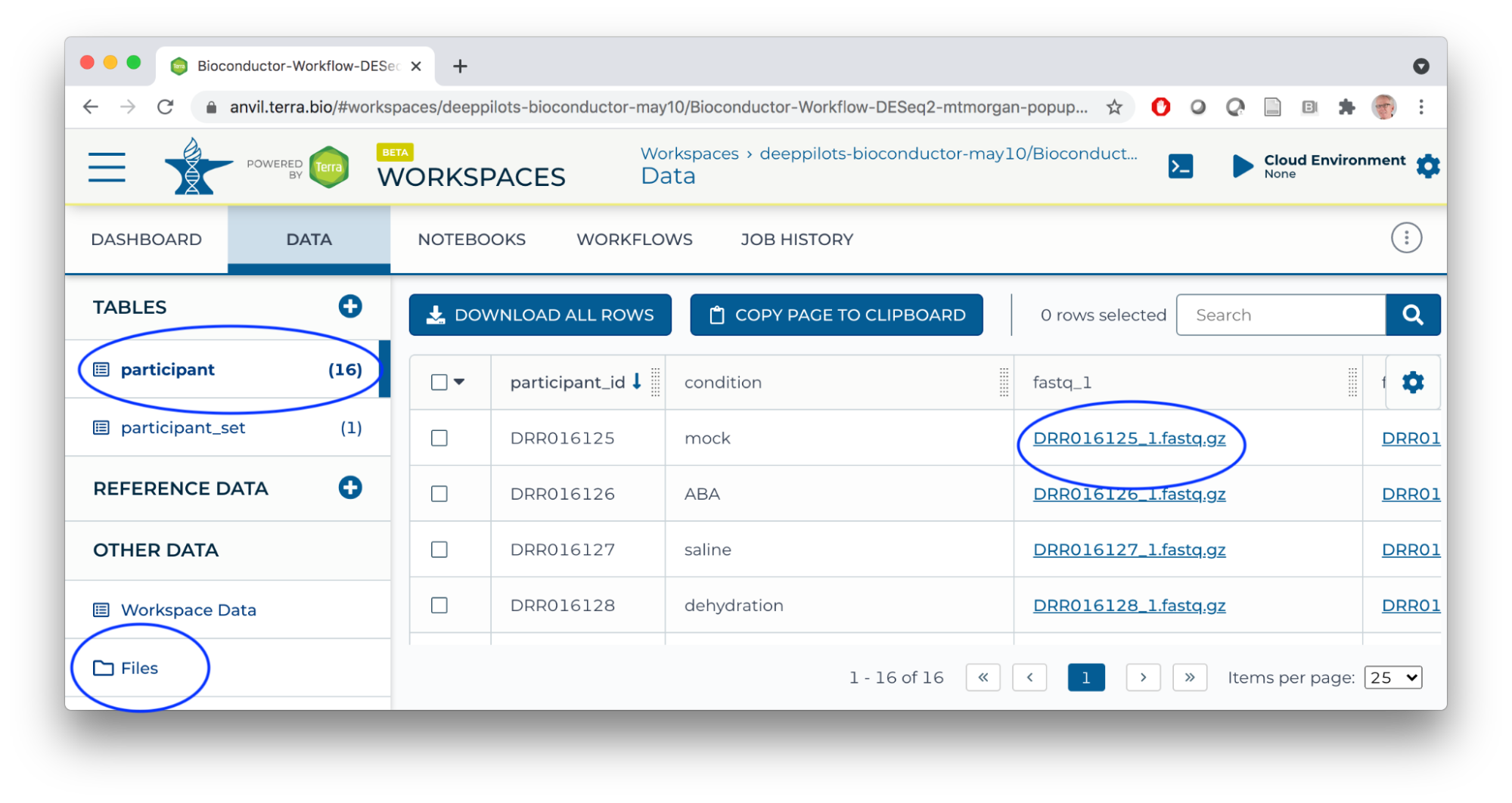
- DATA
- participant, participant_set TABLES
- Files in google bucket contain only notebooks
- NOTEBOOK
- How to use all aspects of the workspace for bulk RNASeq differential expression analysis.
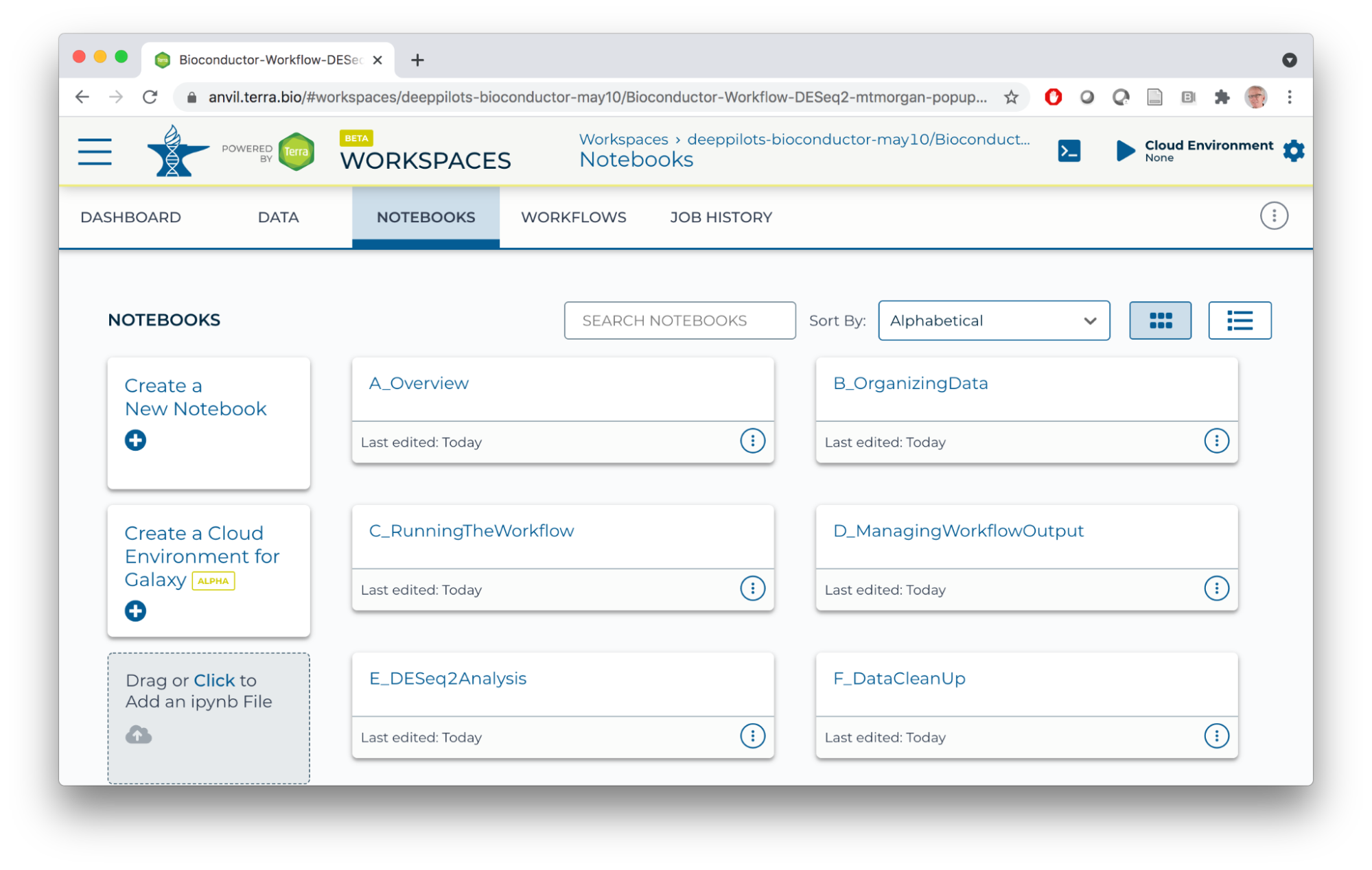
- How to use all aspects of the workspace for bulk RNASeq differential expression analysis.
- WORKFLOW
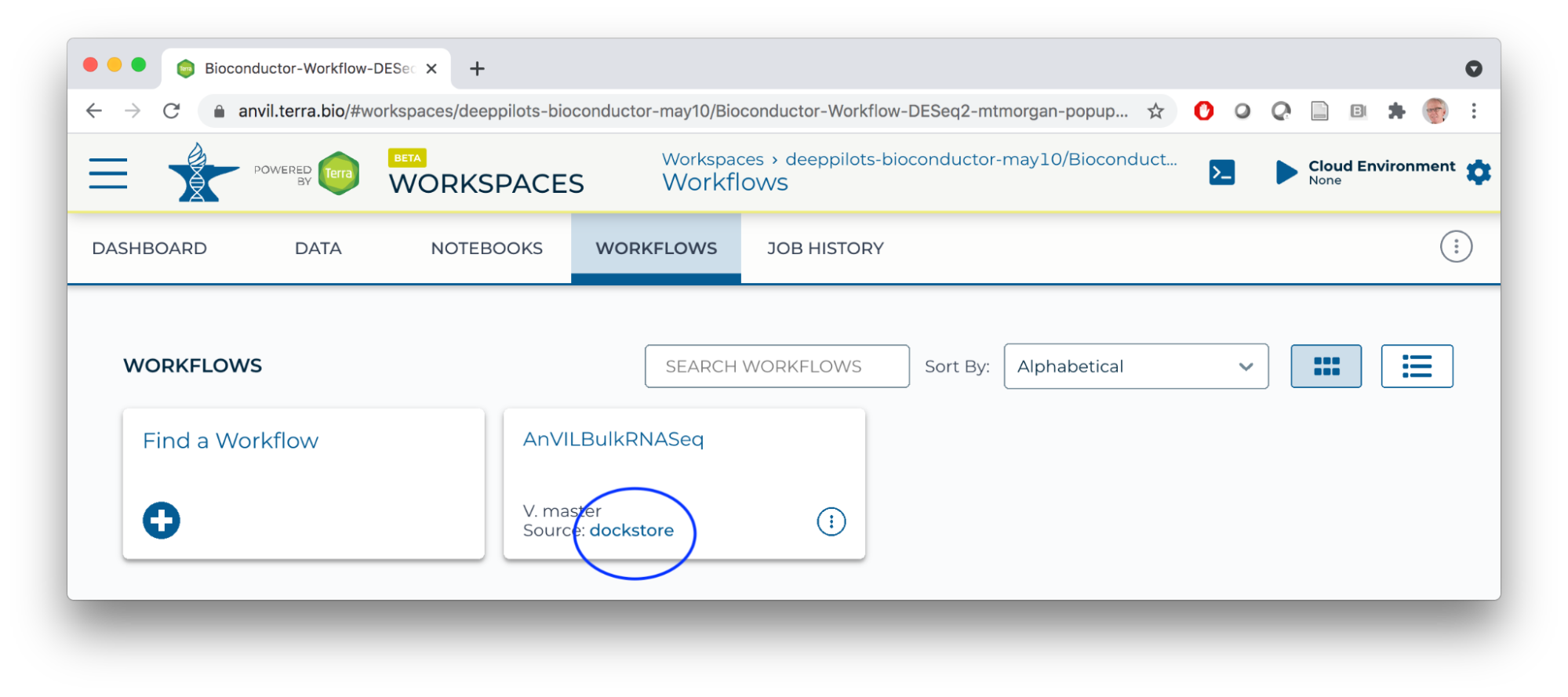
Running a Workflow
salmon quantification
- Inputs
- Transcriptome FASTA file
- Per-sample paired-end FASTQ files
- Outputs
- Per-sample counts of reads aligned to known transcripts
- 'Ultra fast' aligner -- should take about 20 minutes for the largest FASTQ file
Launch
- SELECT DATA from participant_set
- Connect workflow INPUTS to columns in the participant table (FASTQ files), workspace bucket (transcriptome FASTA file), or direct entry (transcriptome name)
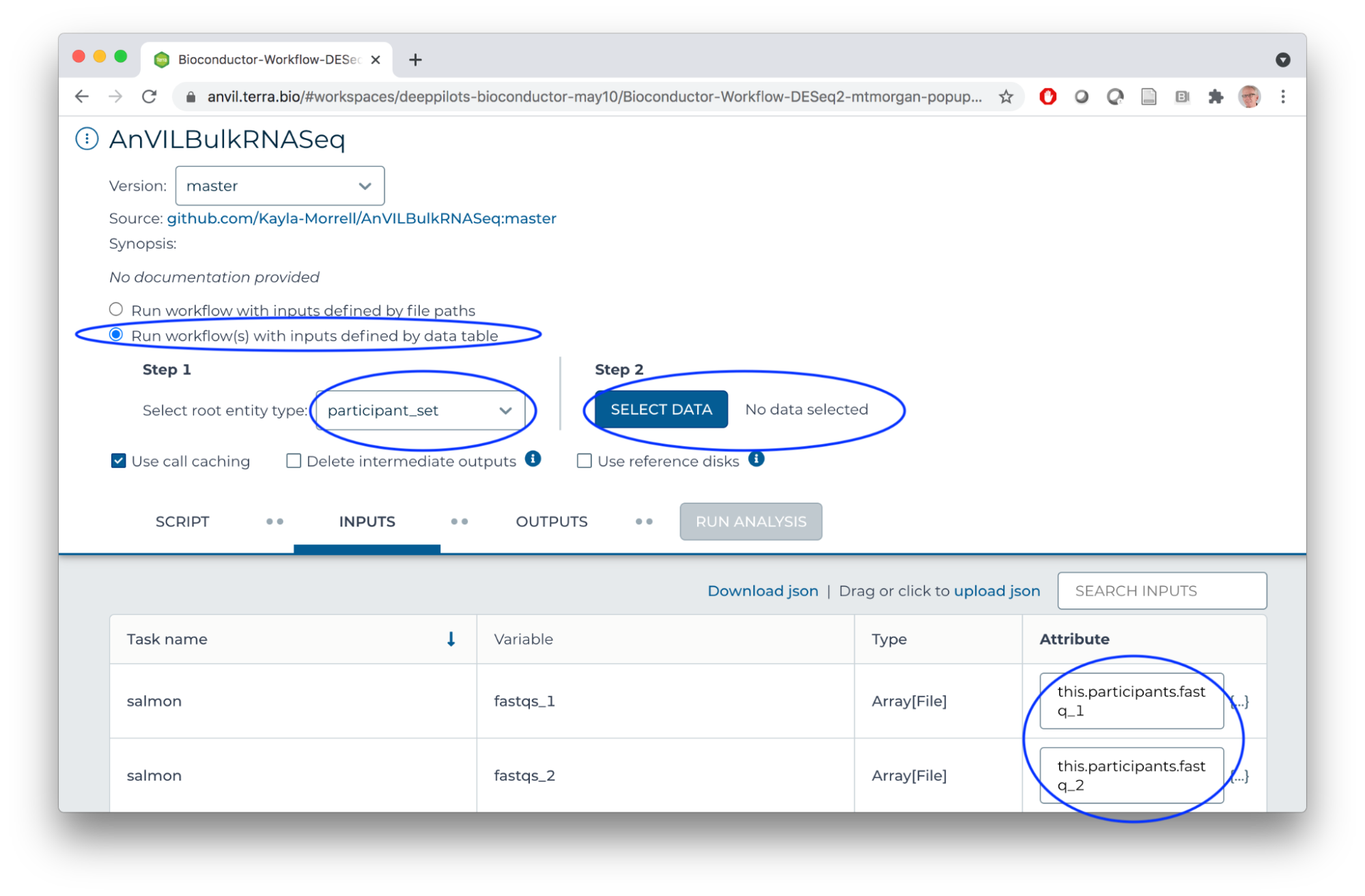
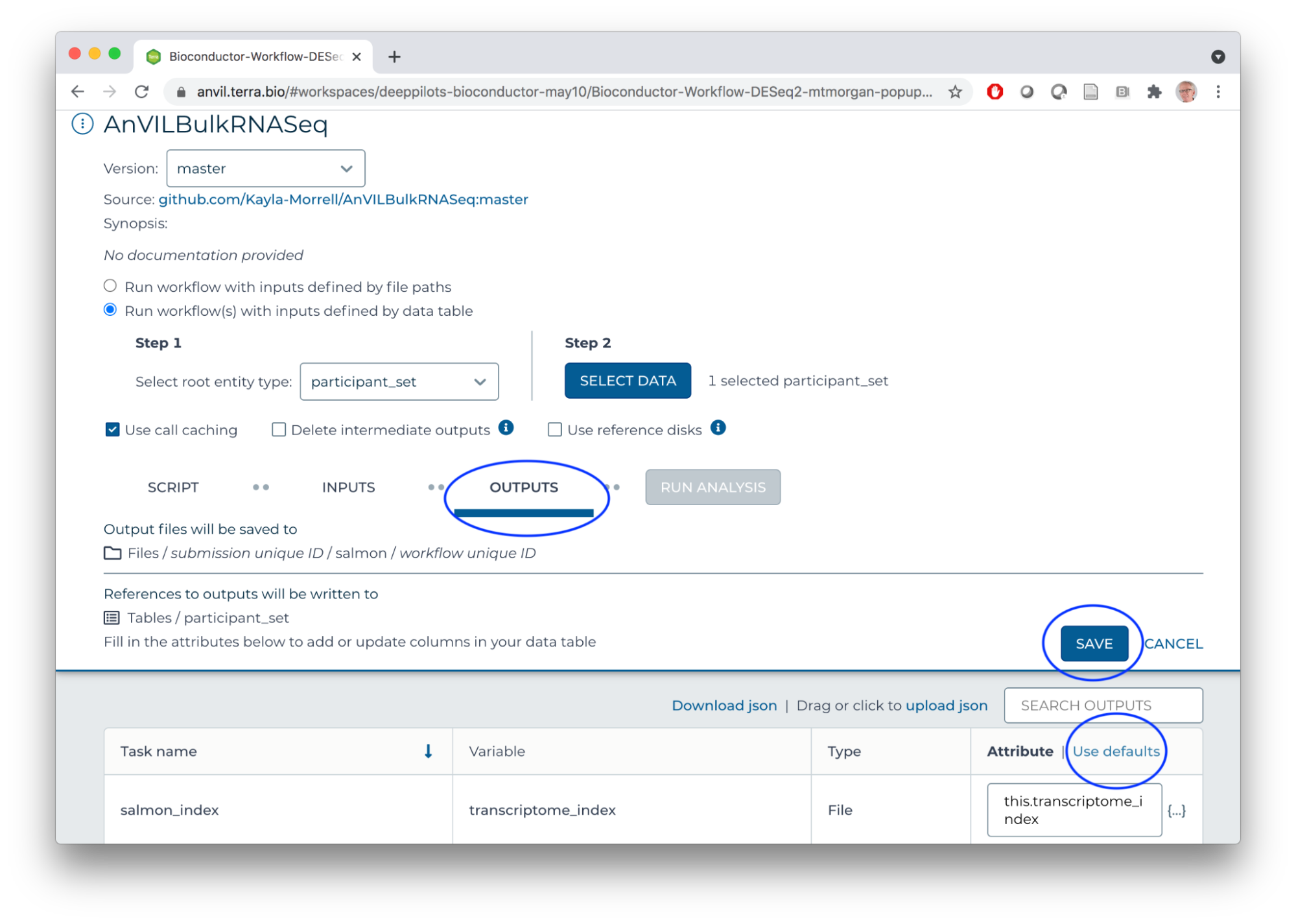
- Use default OUTPUTS
- SAVE
- RUN ANALYSIS
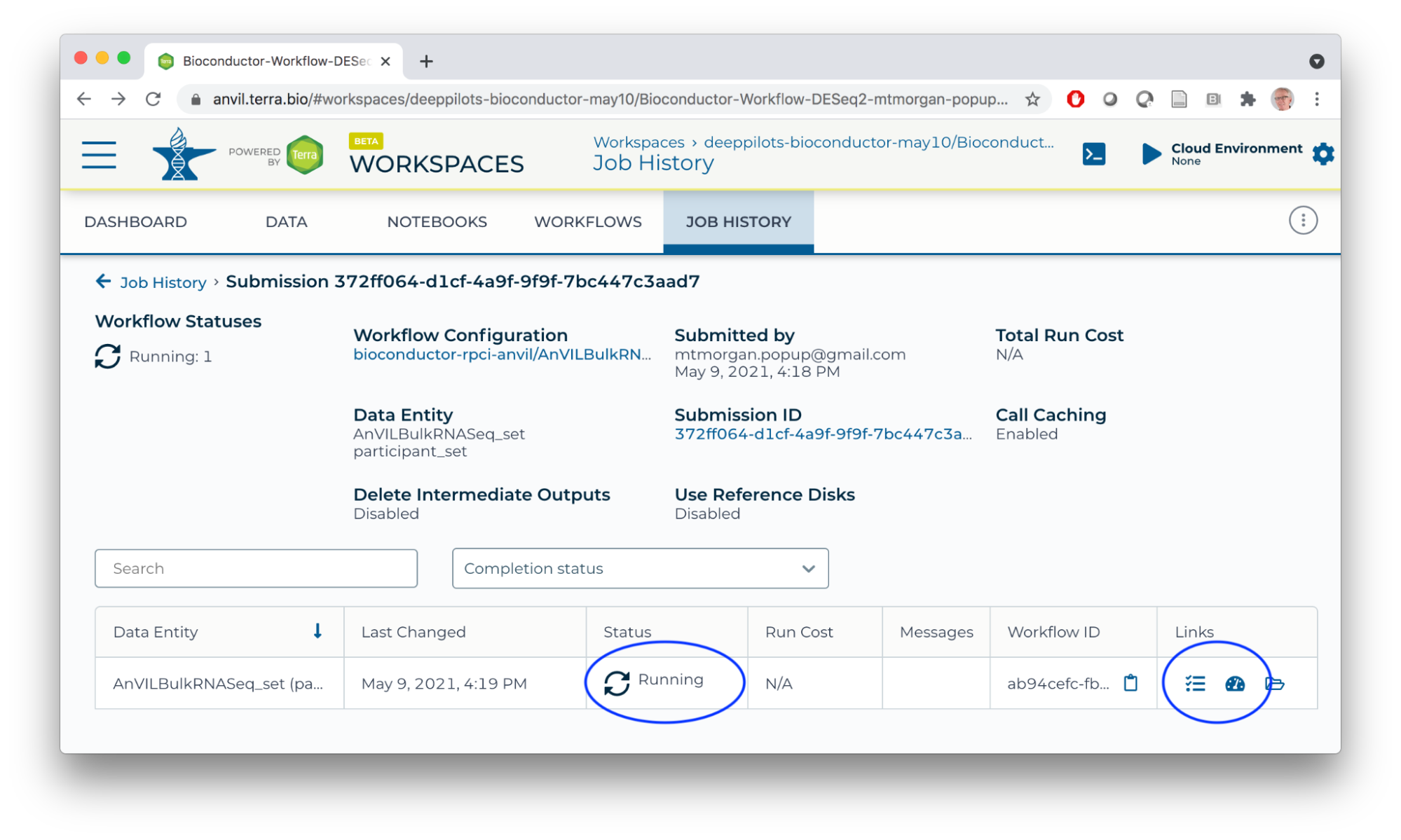
Monitor
Runtime
- In a new tab... some preliminary work, necessary until the image is updated in the next 2 months.
- Start a Jupyter Notebook interactive environment
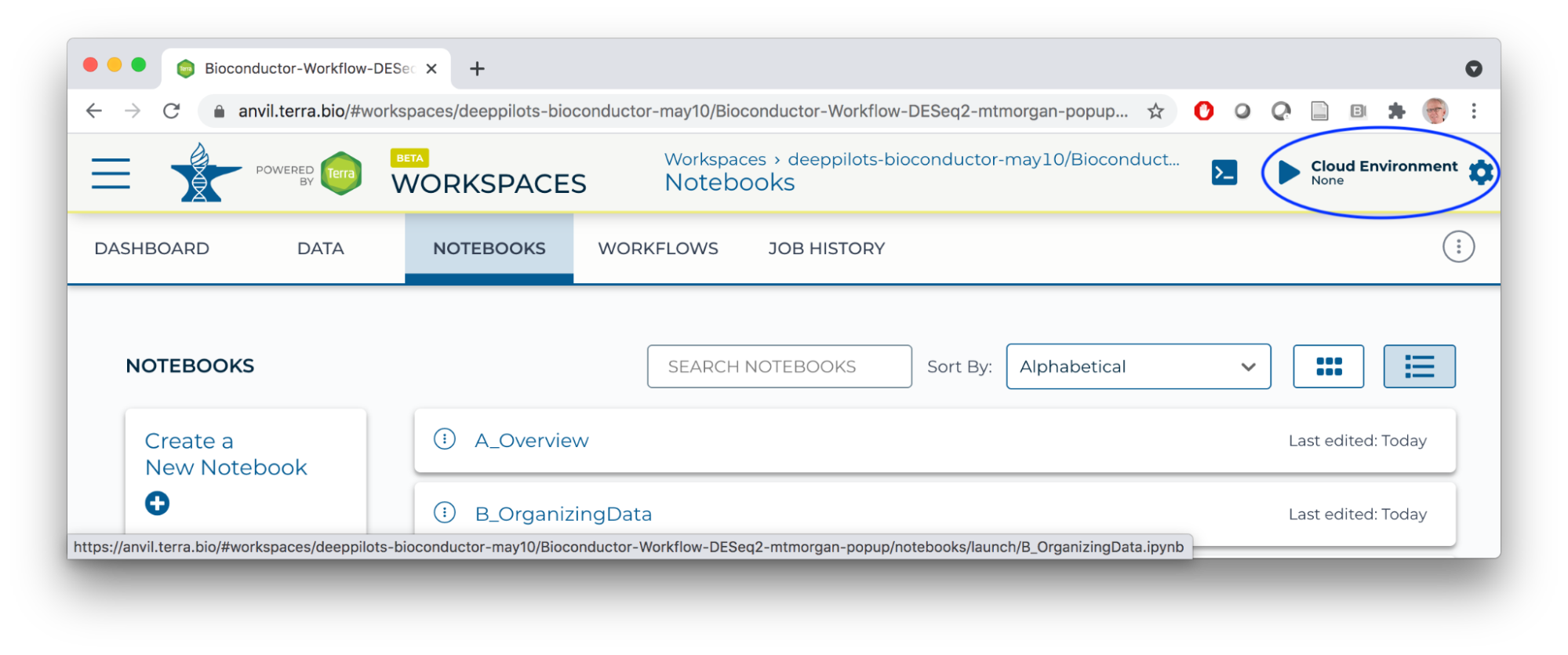

- When the runtime is ready, launch an interactive shell to update packages

- An interactive shell is 'better' for updating packages because we can see progress / errors; these are hidden by the Jupyter notebook
- Start R, update installed packages, and install current version of the AnVIL package
root@...> R options(Ncpus = 2) # faster installation, even if runtime 'oversubscribed' BiocManager::install(ask = FALSE) # update installed packages pkgs <- c("Bioconductor/AnVIL", "GenomicFeatures", "tximport", "DESeq2") BiocManager::install(pkgs) # latest AnVIL package
Workflow Components
WDL
- Available through Dockstore, from GitHub salmon.wdl
task- Logical collection of tasks on a homogeneous input, e.g., 'align FASTQ files of one sample'
runtime: e.g., docker image; could also specify memory, CPU, disk size, etc
workflow- Collection of tasks into an overall execution sequence
scatter- Distribute a vector of operations (e.g., FASTQ file pairs from each sample) across compute nodes
Customizing analysis
- Make your FASTQ files and transcriptome FASTA files available in the cloud
- Update the participant and participant_set DATA TABLE with relevant information about your experiment, including links to the FASTQ files of each sample. Use the AnVIL package to help accomplish this.
- Run the workflow on the new data!
Developing workflows
- Local development
- Use Cromwell (Java application) to run locally.
- Develop individual tasks on small subsets of data.
- Transition to the cloud for large scale testing / full workflow.
- Dockstore / GitHub
Interactive Analysis
Setup
- Notebooks A, B, and C describe how the workspace was set up; review at your leisure. This material may be useful when running on your own data.
Notebook D_ManagingWorkflowOutput
- Extracts relevant files from the workflow output to the local disk
- Open in 'EDIT' mode
- Enter each evaluation cell and press return
Notebook E_DESeq2Analysis
- Creates an object suitable for use in the DESeq2 vignette.
Summary
What You've Accomplished
Running a Workflow
- Bulk RNASeq analysis from FASTQ files to 'top table' for interactive analysis.
- Relationship between DATA TABLEs, workspace bucket, and workflow inputs and outputs.
- Launch and monitor workflow progress.
Workflow Components
- Brief introduction to WDL task and workflow components, scatter operation, and runtime environments.
- Steps to developing your own workflow using local execution on small example data.
Interactive Analysis
- Launch and use Jupyter notebooks.
- Workflow output retrieval from workspace bucket to local disk.
- Management of data for input to DESeq2.
Next Steps
- Follow instructions at Set up billing with $300 Google credits to explore Terra to enable billing for your own projects.
Frequently Asked Questions
-
(From Liz) This article discusses the differences between your Cloud Environment VM and the one that is created when you run workflows: Understanding the Terra ecosystem and how your files live in it and it links to this article on What happens when you launch a workflow? The articles also discuss where your files are.
-
(From a participant) "Got error at step E: Error in system(paste(which, shQuote(names[i])), intern = TRUE, ignore.stderr = TRUE): cannot popen ‘/usr/bin/which ‘gcloud’ 2>/dev/null’, probable reason ‘Cannot allocate memory’. Anyway to fix it? Thanks."
Superficially, this sounds like you ran out of memory on your runtime. A first step might be to restart the 'kernel' in the Jupyter notebook via one of the menu items. This would mean that you'd have to re-do all computations in the notebook that the cell you're going to evaluate depends on. Another solution might be to restart the 'Cloud Environment' but with more than the default, very modest, 3.75 GB. But actually, this seems like an unusual error and it would be great to understand what perhaps unusual steps you took to get into this situation.