- Guides and Tutorials
- Bioconductor / RStudio
- Starting RStudio
- Using R / Bioconductor in AnVIL
- The R / Bioconductor AnVIL Package
- Running a Workflow
- Single-cell RNASeq with 'Orchestrating Single Cell Analysis' in R / Bioconductor
- Using AnVIL for teaching R / Bioconductor
- Reproducible Research with AnVILPublish
- Participant Stories
- Galaxy
- Starting Galaxy
- Jupyter
- Starting Jupyter
The R / Bioconductor AnVIL Package
Martin Morgan, Nitesh Turaga
An exploration of how workspaces provide a framework for managing data and large-scale analyses using the HCA Optimus Pipeline and 1000G-high-coverage-2019 workspaces and R using the AnVIL package.
Learning Objectives
This week we'll explore how workspaces provide a framework for managing data and large-scale analyses. We use the HCA Optimus Pipeline and 1000G-high-coverage-2019 package.
Key Resources
- Visit https://anvil.terra.bio to use the AnVIL platform.
- We use week-2-demo.R to guide us through this workshop.
- We use the HCA Optimus Pipeline and 1000G-high-coverage-2019 workspaces as examples.
- Review the Introduction to the AnVIL package vignette.
Review
Previously...
- Notes and recorded session: Using R / Bioconductor in AnVIL
Essential Steps
- Login
- Workspaces
- Billing accounts
- Cloud environment -- (R-based) Jupyter notebooks or RStudio
Cloud Computing Environment
- Runtime and persistent disk
- A 'personal' cloud computing environment
- Not shared with others
- Ephemeral
FAQs
- Persistent disk mounted at
- R / Jupyter:
/home/jupyter-user/notebooks - RStudio:
/home/rstudio
- R / Jupyter:
- Startup script or custom docker file for 'sudo'-like access, and for complete reproducibility
Workshop Activities
Setup
- Log in to AnVIL using the email address you used to register for the course and navigate (via the HAMBURGER) to Workspaces.
- If you cloned the Bioconductor-Workshop-Popup workspace last week, delete it now.
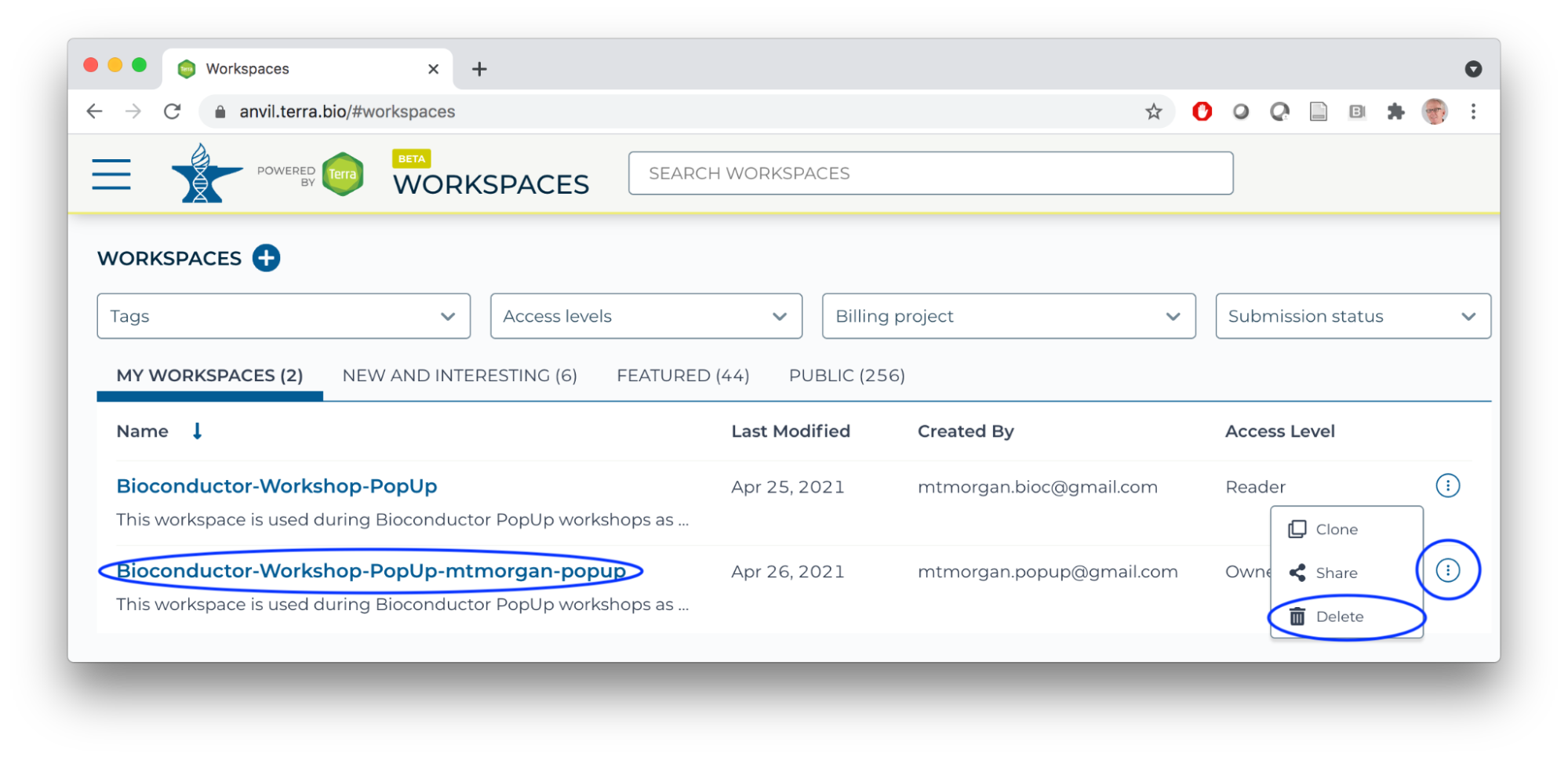
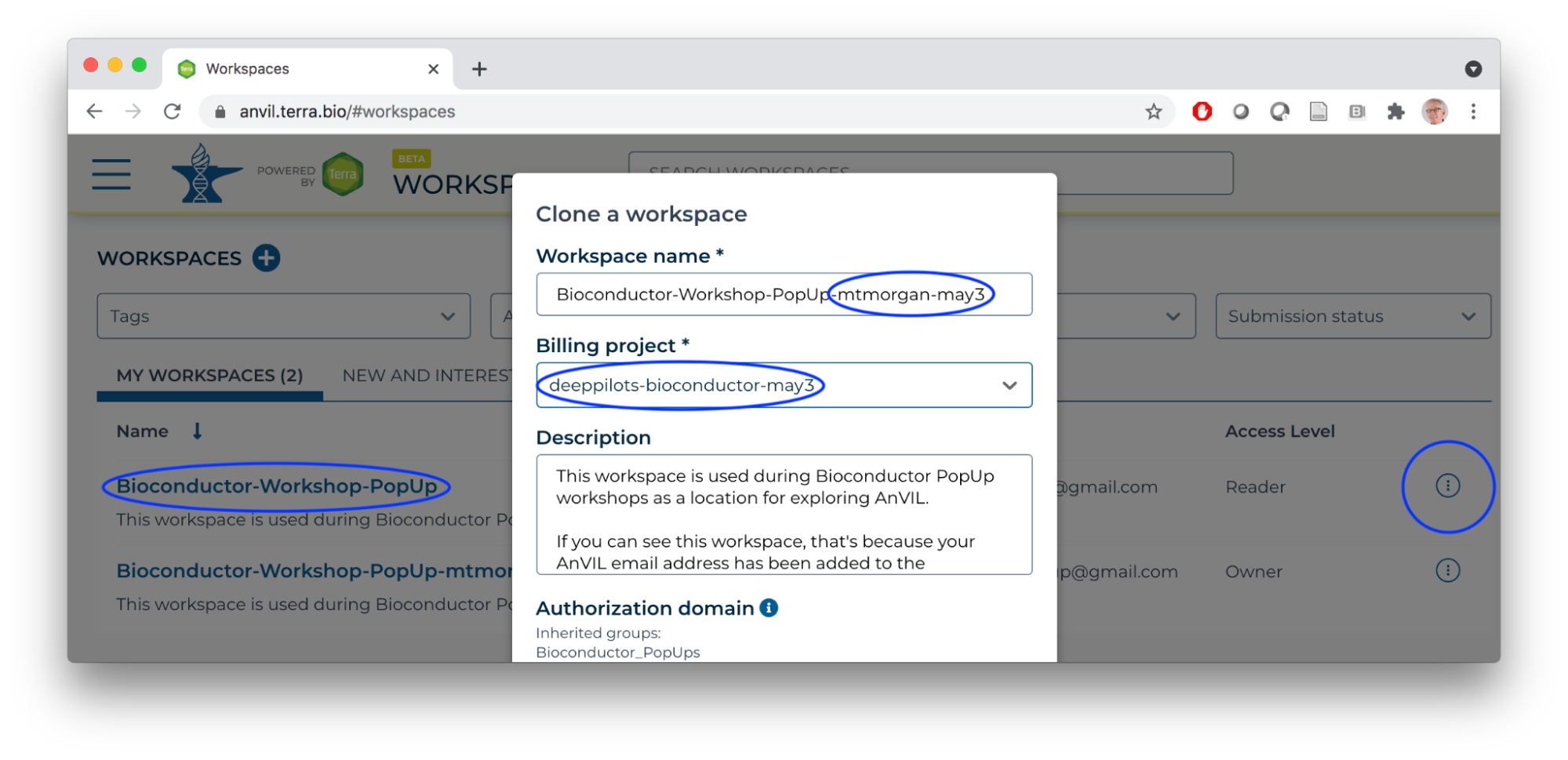
- Clone the Bioconductor-Workshop-Popup.
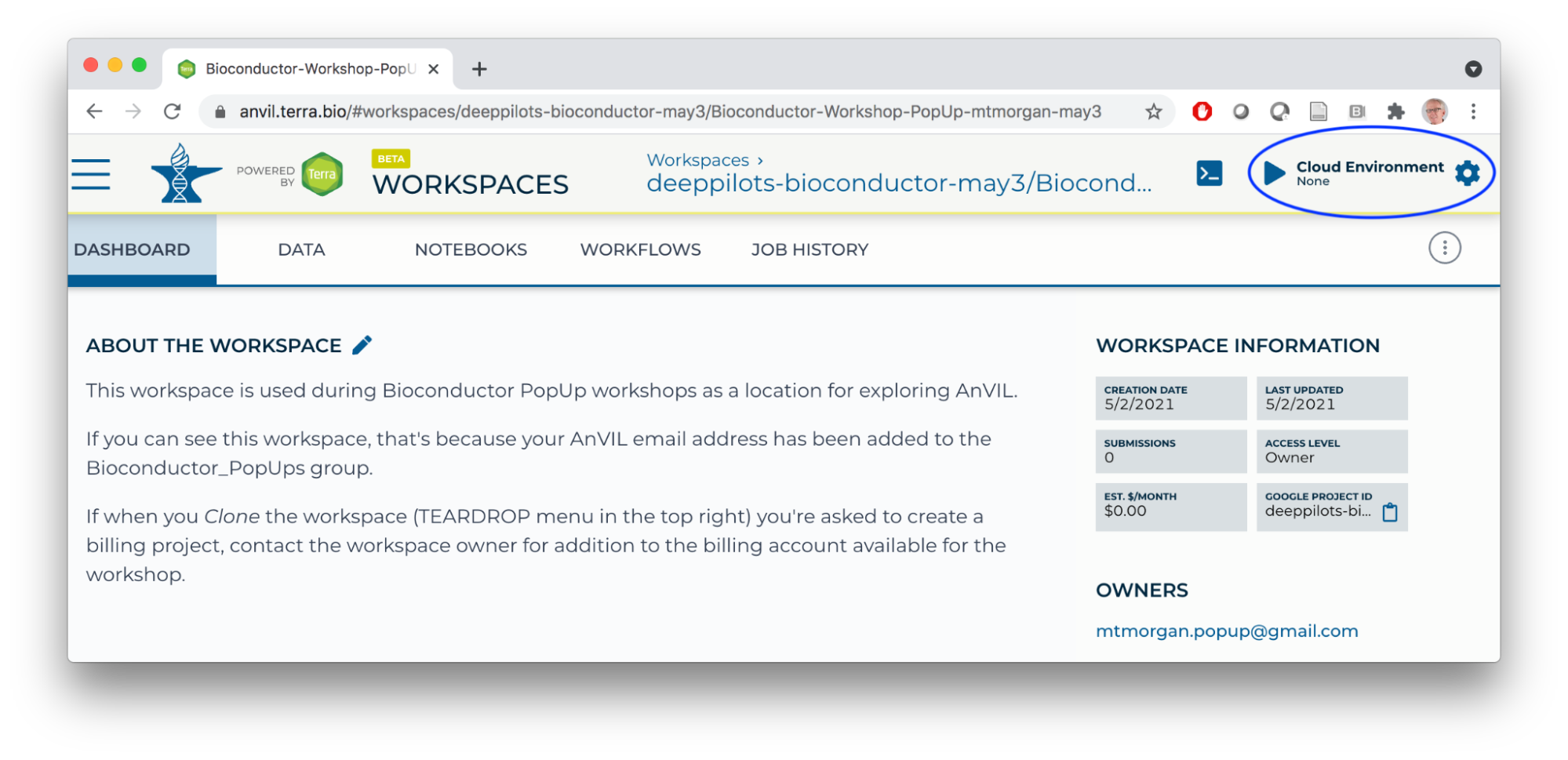
- Start an RStudio cloud environment.
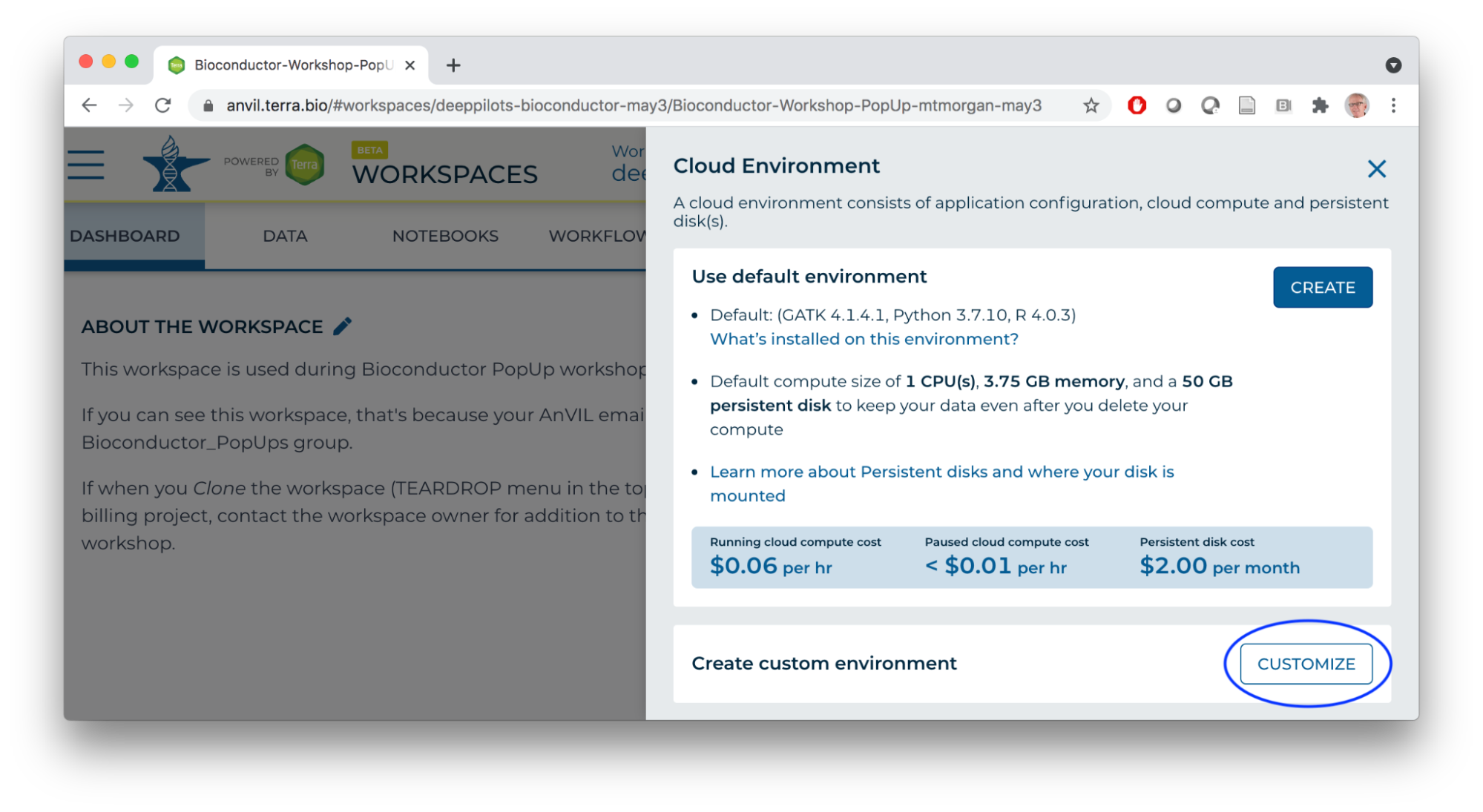
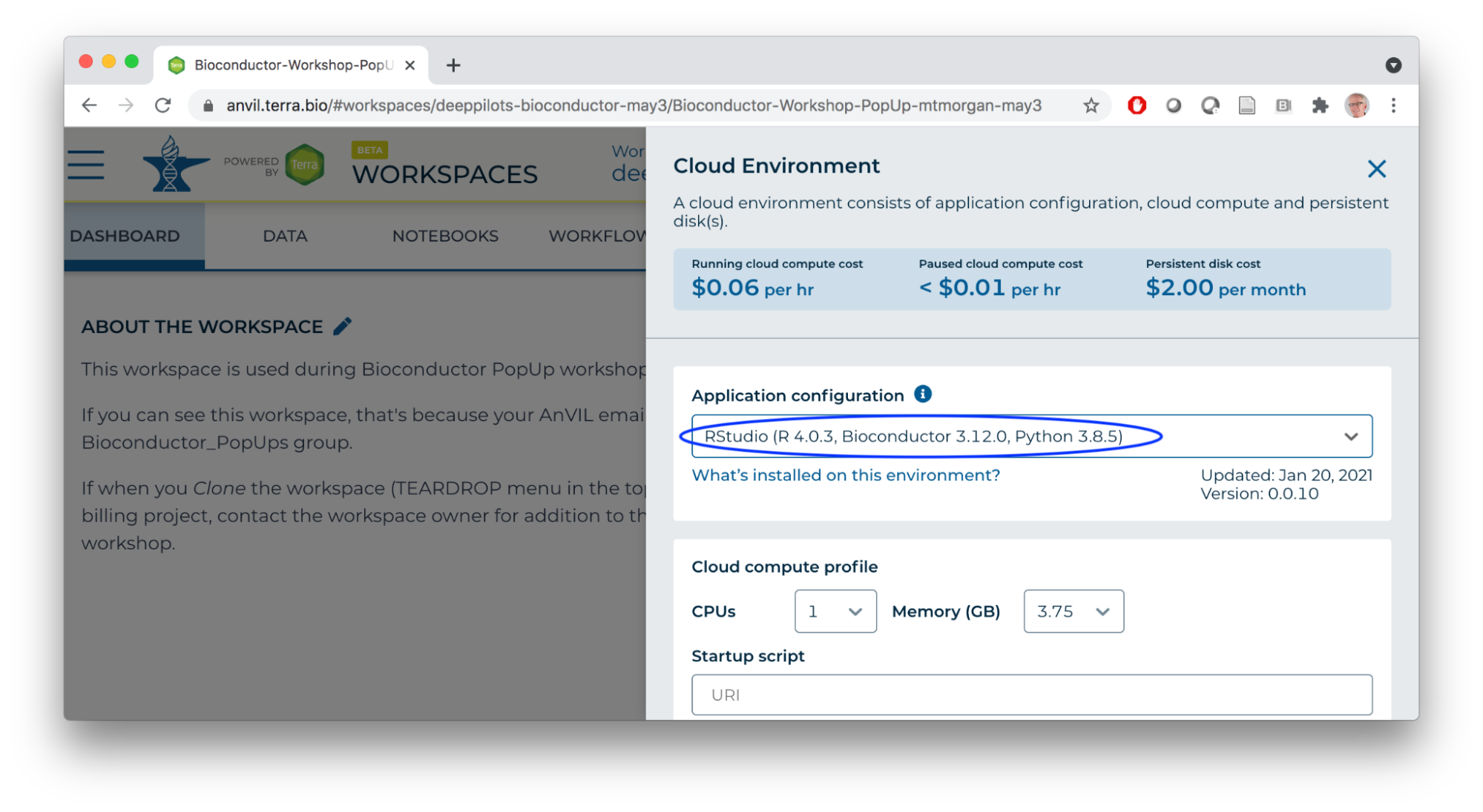
- Launch the cloud environment.
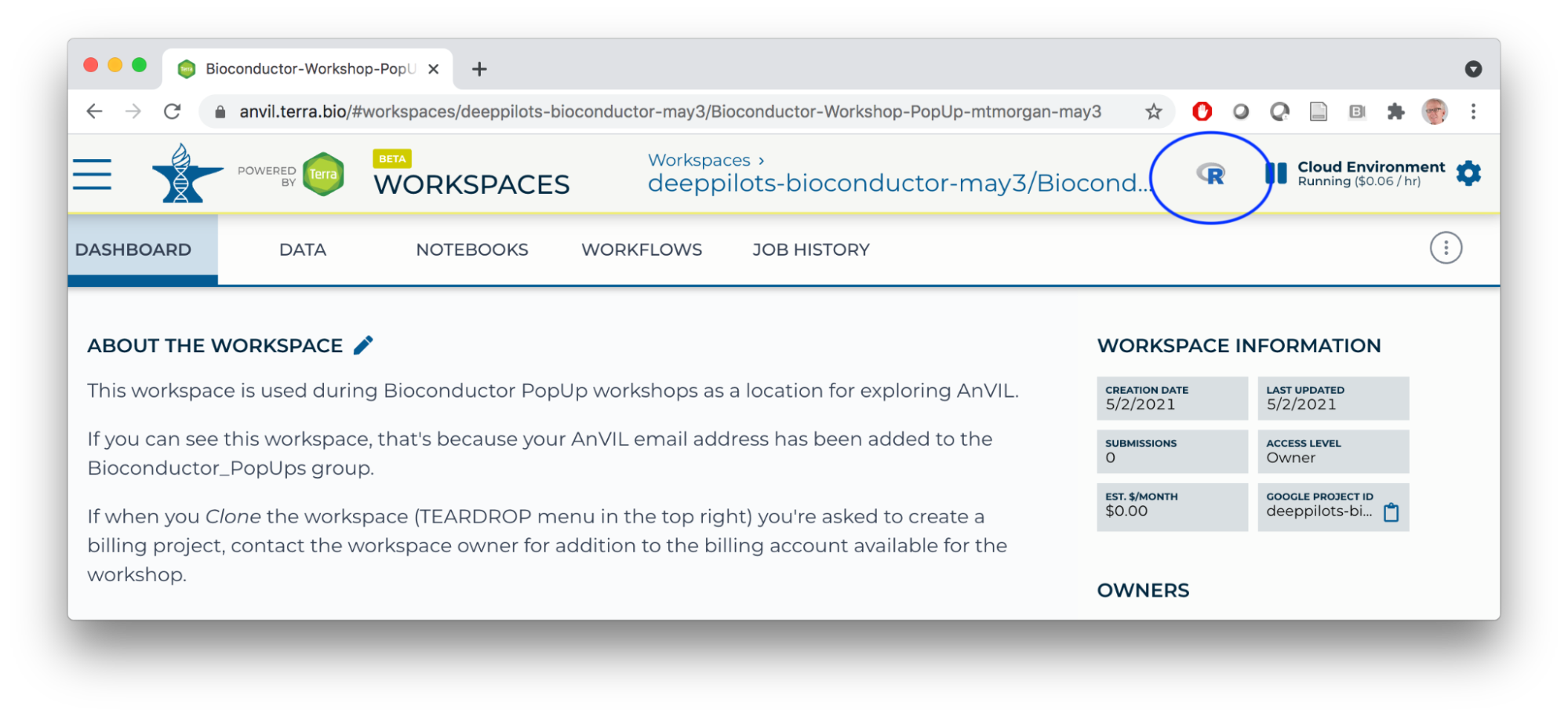
- Copy the week-2-demo.R script into a file on your cloud environment.
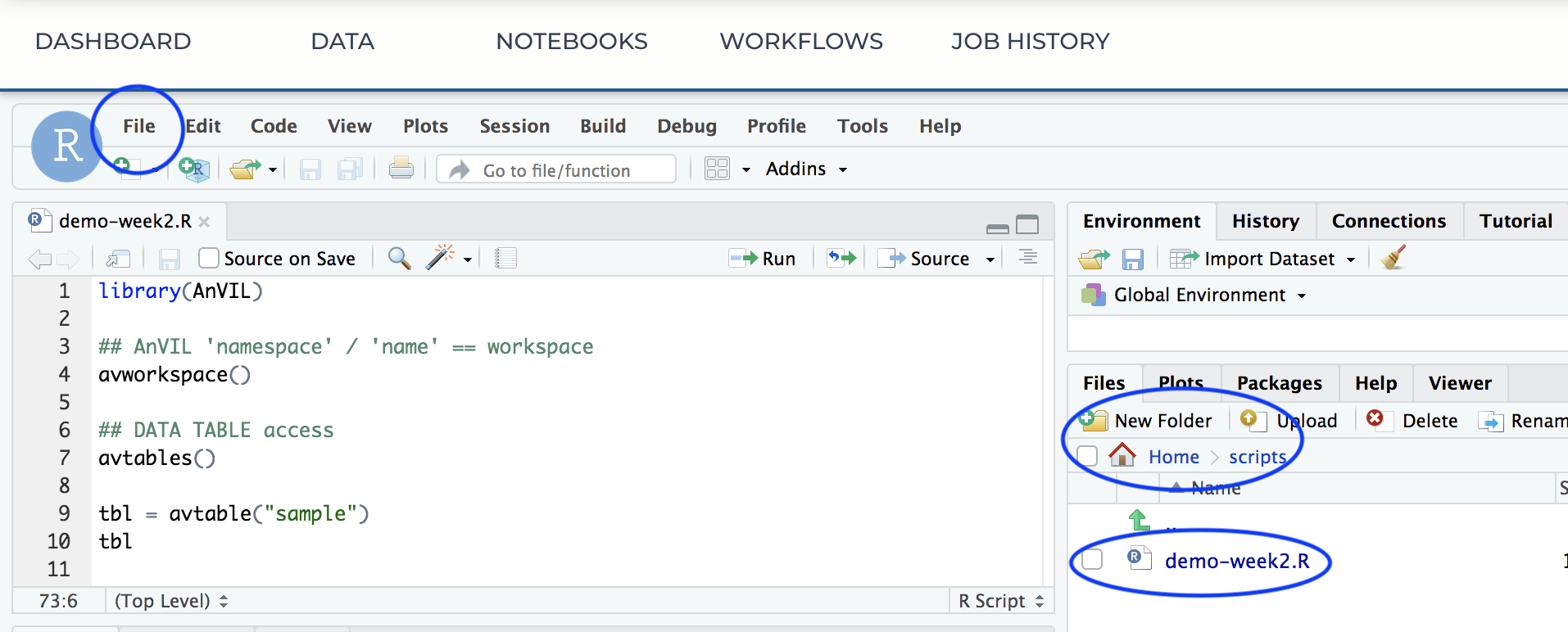
Workflows
- In a new browser tab/window, navigate (via the HAMBURGER) to the HCA Optimus Pipeline workspace. This workspace demonstrates how scRNA-seq fastq files can be transformed to a 'count matrix' for interactive analysis.
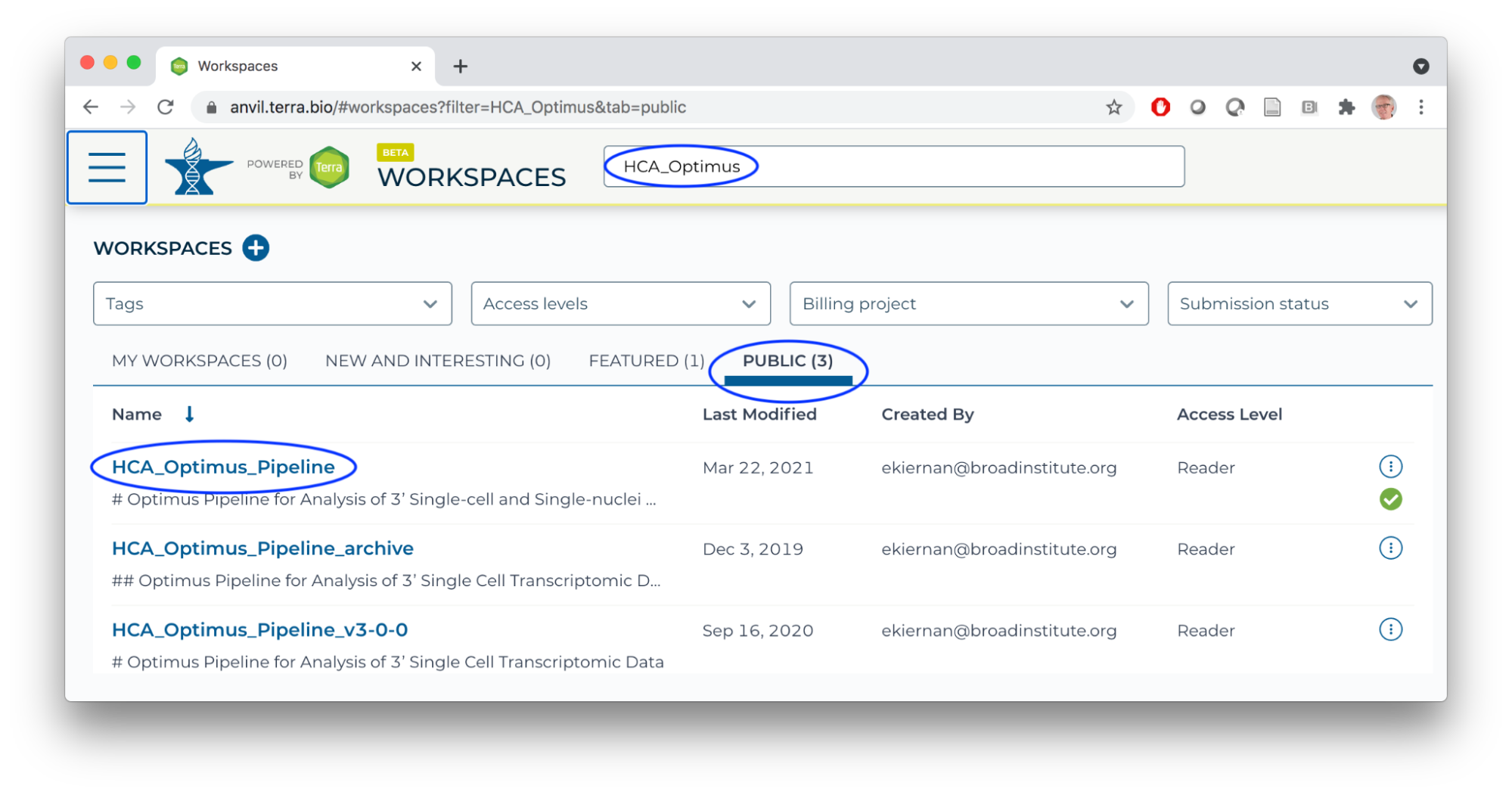
- Overall orientation: DATA TABLES serve as input to WORKFLOWS (scalable 'big data' computation).
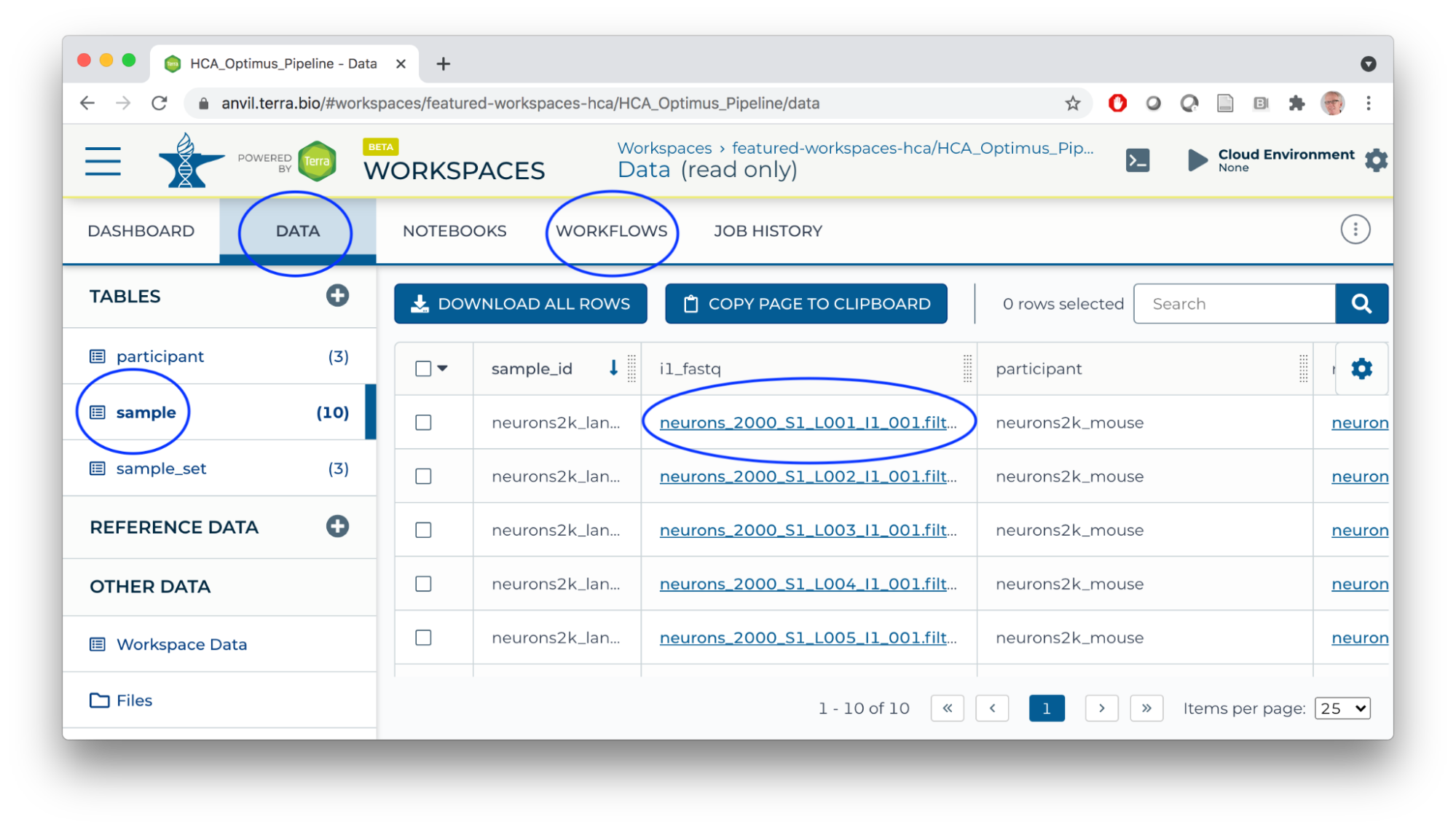
- Workflows transform big data using 'Workflow Description Language' scripts producing outputs (logs, results).
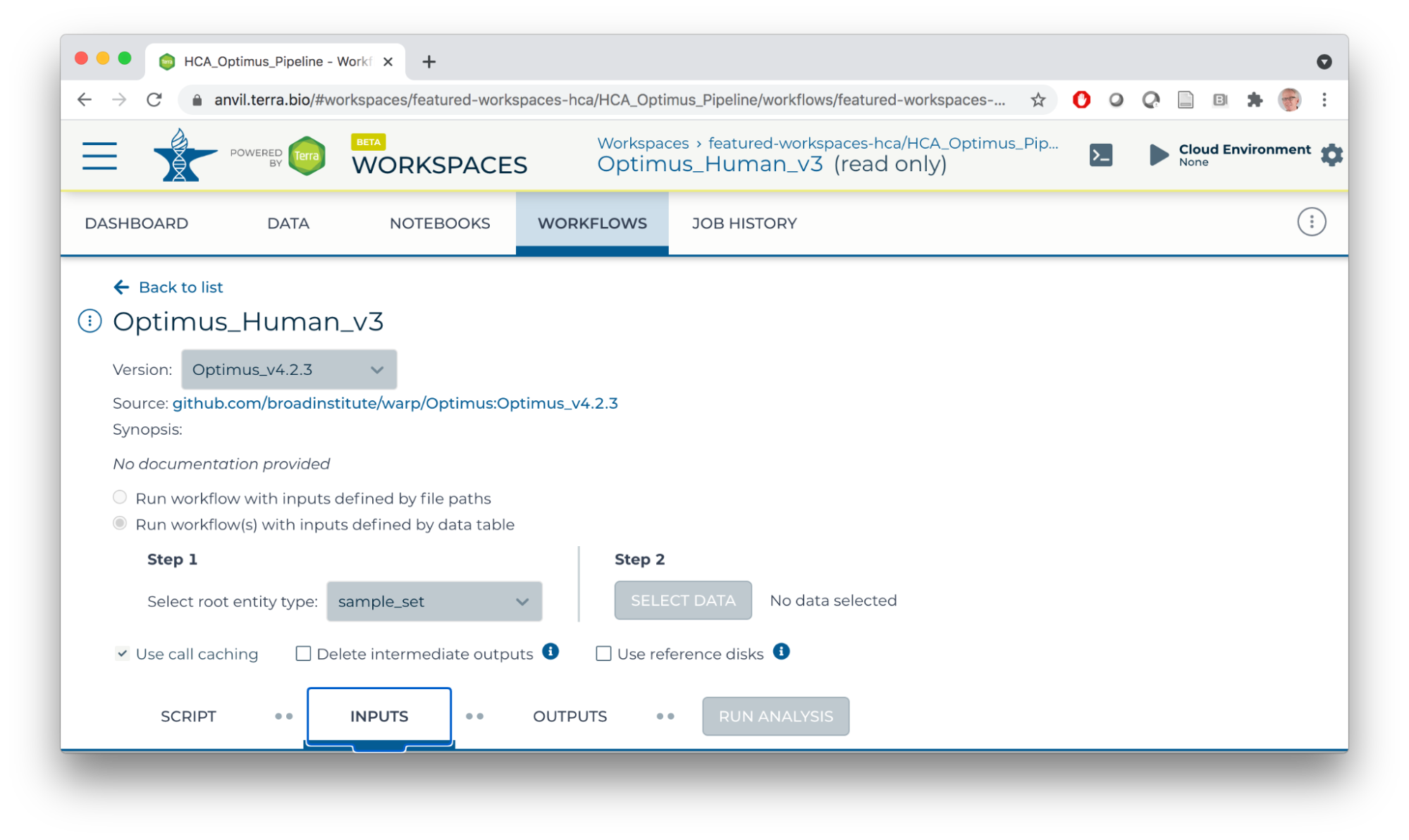 For this workflow:
For this workflow:
- Single-cell RNA seq analysis.
- Inputs are fastq files from individual samples.
- Scripts perform alignment, UMI processing, creating a 'count' matrix of gene x cell (sample) expression matrices, etc.
- Primary output of interest is a 'loom' file summarizing the count matrix.
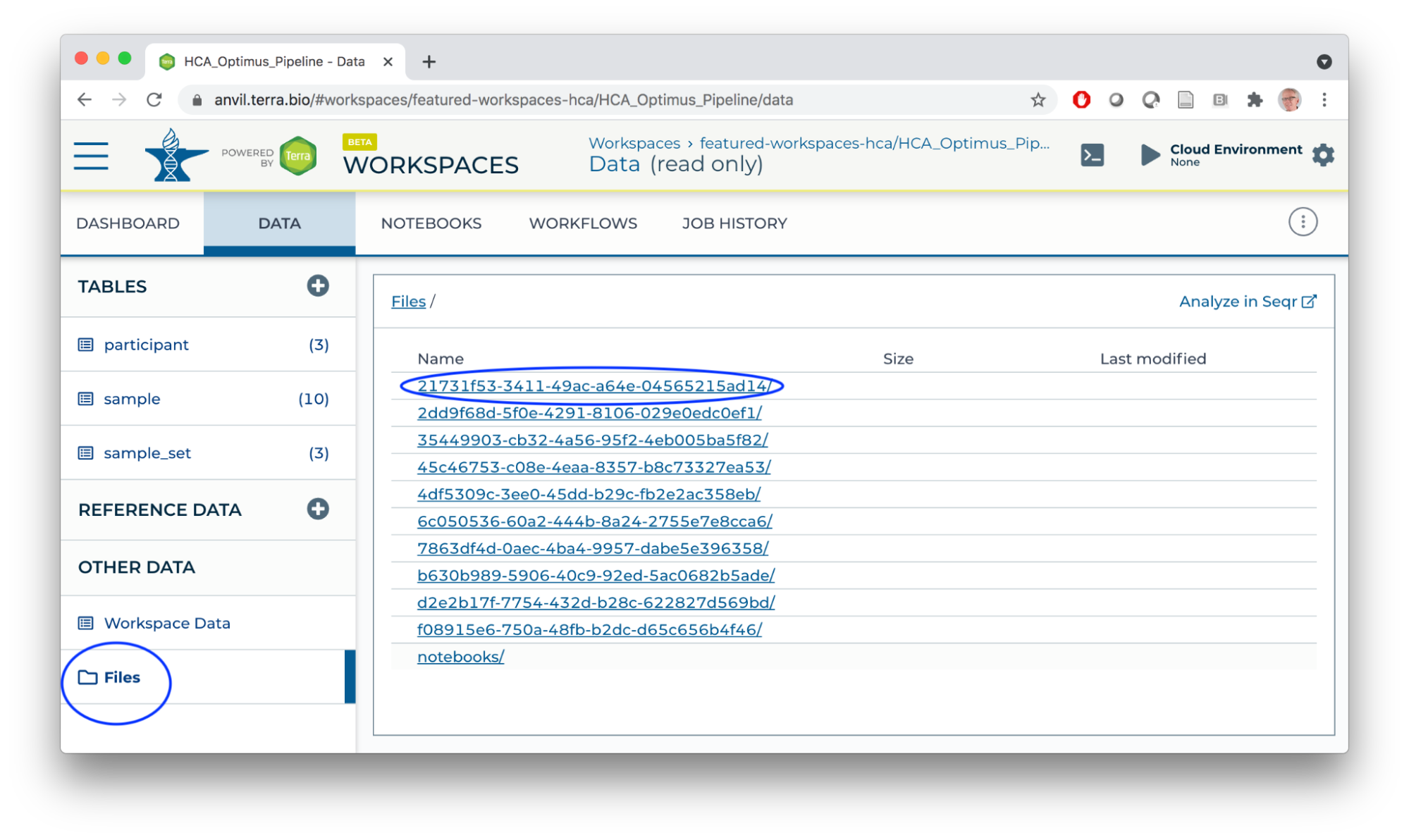
- Workspace bucket / Files store workflow outputs (each workflow run has a unique identifier; logs and results are located under the identifier). Buckets also provide a location for storing and sharing interactive analysis results.
The AnVIL Package
AnVIL Workspaces
hca = "featured-workspaces-hca/HCA_Optimus_Pipeline"
thousand_genomes = "anvil-datastorage/1000G-high-coverage-2019"
library(AnVIL)
avworkspace() # current workspace
avworkspace(hca) # set to HCA workspace
DATA TABLE Access
avtables()
tbl = avtable("sample")
tbl
tbl %>% count(participant)
## tbl %>% avtable_import()
avworkspace(thousand_genomes)
avtables()
participant = avtable("participant")
participant
participant %>% count(POPULATION, sort = TRUE)
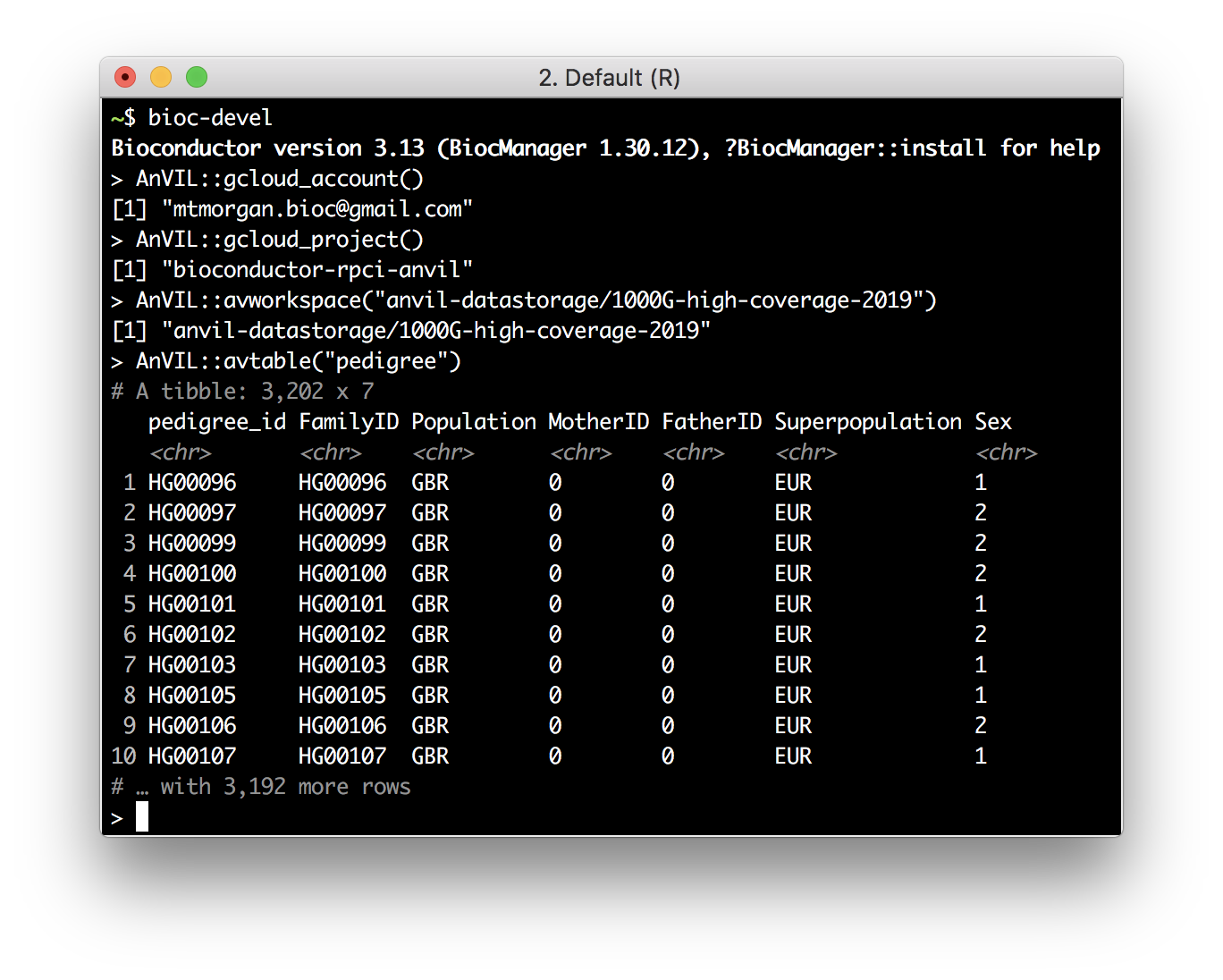
avtable("pedigree") %>%
count(Population, Sex) %>%
tidyr::pivot_wider(names_from = "Sex", values_from = "n")
## switch back to this workspace
avworkspace(hca)
Google buckets
## Copy files from google buckets to persistent disk
tbl = avtable("sample_set")
tbl
dir.create("~/loom")
gsutil_cp(tbl$loom_output_file, "~/loom/") # see also gsutil_rsync()
dir("~/loom")
## Workspace Bucket -- 'backup' or share persistent disk to workspace bucket
avbucket() # bucket associated with this workspace
gsutil_ls(avbucket())
avfiles_backup("~/scripts", recursive = TRUE) # see also avfiles_restore()
gsutil_ls(avbucket(), recursive = TRUE)
Fast Binary Package Installation
## do NOT update out-of-date packages yet
BiocManager::install("Bioconductor/AnVIL")
## RESTART R
AnVIL::repositories() # binary Bioconductor and CRAN package installation
## install and use LoomExperiment
AnVIL::install("LoomExperiment") # about 40 seconds, rather than 10's of minutes
sce = LoomExperiment::import("~/loom/pbmc_human_v3.loom")
Access AnVIL from Outside AnVIL
- Requires gcloud SDK installed on your computer.
- Use SDK to register your Gmail account and google billing project.
Access the AnVIL 'API'
leo = Leonardo()
leo
leo$listDisks()
terra = Terra()
tags(terra, "Workspaces")
wkspc =
terra$listWorkspaces() %>%
flatten() %>%
select(-starts_with("workspace.attributes"))
wkspc
Summary
What You've Accomplished
Setup
- Clone a workspace, launch an RStudio cloud environment
- Navigate between workspaces
Workflows
- Elements of workflow structure -- DATA TABLE inputs, scripts, File outputs
AnVIL Package
- Selecting workspaces
- Managing DATA TABLEs
- Moving data to and from google buckets
- Fast binary package installation (in the 'devel' version of the package)
- Advanced features, e.g., local use, API access
Next Steps
- Follow instructions at Set up billing with $300 Google credits to explore Terra to enable billing for your own projects.
Frequently Asked Questions
- Uploading workflows -- through GitHub / Dockstore, but also the Broad Methods Repository (YouTube); see also the WDL Puzzles workspace.
- Default name and namespace -- the runtime starts in a particular workspace, and the runtime knows the default namespace and name. So by default, I had
> avworkspace() [1] "deeppilots-bioconductor-may3/Bioconductor-Workshop-PopUp-mtmorgan" gsutil_cp(): CommandException: Downloading this composite object requires integrity checking with CRC32c, but your crcmod installation isn’t using...This is a bug that should be fixed in the underlying image for the runtime.
Help us make these docs great!
All AnVIL docs are open source. See something that’s wrong or unclear? Submit a pull request.
Make a contribution